Admixture in a given modern/ancient population or individual (as opposed to uniparental haplogroup analysis) can be determined using various methods. Most methods estimate admixture proportions directly, whether it be through the use of arbitrary genetic populations or specific source populations. The genetic structure of a population or individual can also be inferred using a PCA (principal component analysis), which can allow for the visualization of a population's admixture.
One of the programs that is frequently used when analyzing admixture is the suitably-named ADMIXTURE. ADMIXTURE is a program developed by David H Alexander, John Novembre, and Kenneth Lange for Linux and Mac. It estimates the ancestry of a population using K (a given number) populations. It does so in a model-based manner. It utilizes a given dataset containing samples (which can be either ancient or modern, and can be sequenced in any way), and estimates the amount of genetic ancestry of the samples derived from each of K populations, though it does not model genetic drift. These populations are hypothetical and not designated by the user, meaning that ADMIXTURE does not directly test for admixture between populations, examining individual samples instead of populations as a whole. Admixture proportions are almost invariably displayed via barplots. The program can be accessed here.
STRUCTURE is a program developed by J K Pritchard, M Stephens, and P Donnelly. It uses the same statistical model as ADMIXTURE, but is less efficient than ADMIXTURE. Unlike ADMIXTURE, STRUCTURE is available for Linux, Windows, and Mac. Nonetheless, ADMIXTURE is preferred over STRUCTURE due to the fact that ADMIXTURE uses data formats that are used by other programs, whereas STRUCTURE does not. The program can be accessed here.
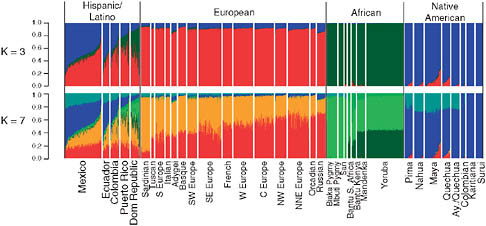
Image courtesy of "Genome-wide Patterns of Population Structure and Admixture Among Hispanic/Latino Populations--Katarzyna Bryc, Christopher Velez, Tatiana Karafet, Andres Moreno-Estrada, Andy Reynolds, Adam Auton, Michael Hammer, Carlos D. Bustamante, and Harry Ostrer." This image demonstrates what ADMIXTURE/STRUCTURE models can look like.
Another software that has been used in a myriad of publications is ADMIXTOOLS. ADMIXTOOLS is a collection of programs that use f-statistics derived from direct genetic data (SNPs) to infer genetic relationships between populations. The original ADMIXTOOLS was developed by David Reich and Nick Patterson; the program was developed for Linux and Mac (the program can be accessed here).
A revised version of the software, ADMIXTOOLS 2 (which can be accessed here), was developed by Robert Maier, Pavel Flegontov, Ulas Isildak, David Reich, and Nick Patterson for Linux, Mac and Windows. ADMIXTOOLS 2 serves to improve upon the original ADMIXTOOLS and utilize more efficient processes for analyzing admixture.
All ADMIXTOOLS programs utilize f-statistics (specifically f2 statistics) as their basis, which describe how populations are related to each other. The programs that comprise ADMIXTOOLS (and ADMIXTOOLS 2) are qpGraph (this program graphs admixture proportions using a "branching" model), f3 (this program can be used to find out if a population is admixed between other populations), f4 (this program can be used to determine whether a group of populations forms a clade/"branch" compared to another group), qpF4ratio (this program can be used to quantify the amount of admixture derived from different populations), qpWave (this program can be used to determine whether a group of populations forms a clade/"branch" compared to another group and to estimate waves of admixture that separate two sets of populations), qpDstat (this program can be used to determine whether populations form clades as well), and qpAdm (this program can be used to quantify the amount of admixture derived from different populations and find out if a population is admixed between other populations).

Image courtesy of Robert Maier. This image displays what a qpGraph can look like.
qpAdm is arguably the most ubiquitous out of any of these programs, and is used by hobbyists and academics alike. qpAdm utilizes f-statistics to determine the admixture proportions of a given target population, and allows the user to ascertain plausible models of admixture. Its primary use is to designate plausible models of admixture for a given target population and eliminate implausible models. The user designates a target population/sample (the population/sample whose admixture will be modeled), up to four source populations (which will be used to model the admixture of the target), and outgroups (technically a misnomer, as qpAdm outgroups should include populations that are differentially related to source and target populations. Outgroups in qpAdm are populations that did not contribute to the ancestry of the target and serve as reference populations compared to the source and target populations, utilizing a "branching" model). The program then elucidates various models with plausibility values that are used to determine whether the value "passes" or "fails". qpAdm can be used on samples that have low genetic coverage, but, as delineated by Harney et al., 2021, qpAdm should not be used for modern target populations, as while qpAdm is typically robust to DNA damage, samples with extreme variations in DNA damage can result in biased models with inaccurate proportions, and this is primarily accentuated when co-analyzing modern and ancient samples together. Also, it should not be used for populations with histories of extended periods of gene flow, as qpAdm assumes that gene flow occurs as a single burst over a short period of time. As a result, models operating under that assumption can be misleading and inaccurate; qpAdm cannot distinguish between models containing singular pulses and those containing continuous gene flow. Additionally, qpAdm should not be used when large numbers of outgroup populations are included, since when large numbers of outgroups are used, the chance of the model being rejected increases significantly even if it is a true model. qpAdm also demonstrates bias when modeling populations with samples sequenced using varying methods, especially if samples were shotgun sequenced. These biases are discussed in a tutorial specific to qpAdm.

Image courtesy of Robert Maier. This image displays what a qpAdm output typically looks like.
Due to the fact that qpAdm cannot be used when both modern and ancient samples are co-examined, LINADMIX was developed by Lily Agranat-Tamir, Shamam Waldman, Naomi Rosen, Benjamin Yakir, Shai Carmi, and Liran Carmel. LINADMIX works in tandem with ADMIXTURE, relying on ADMIXTURE's output. LINADMIX uses a linear regression model and estimates admixture proportions for a target population using ADMIXTURE results of source populations as mixing coefficients and computes a plausibility value to determine whether or not the model is plausible, meaning that it can also be used to designate plausible models. LINADMIX can be used to model modern populations, and can be used in cases of missing data and genetic drift (whereas ADMIXTURE cannot model genetic drift, LINADMIX is robust to it). Although LINADMIX performs better when source populations are highly diverged, genetically similar source populations can still be used. The program can be accessed here.
PCAs are used to indirectly infer a population's admixture. PCAs reduce the various dimensions of an individual's genetic data to a few dimensions. A sample will be reduced to coordinates on a plane with the axes representing a principal component, which in the case of population genetics would represent a genetic signature or "shift". While PCAs can be used to infer the admixture of a population as a whole by plotting multiple individuals of a certain population, A sample's coordinates depend on the principal components being used, though the principal components are arbitrary and not designated. These coordinates can then be graphed, allowing the user to visualize various genetic clines and clusters and determine how different populations are related to each other.
One of the most common programs used to generate PCAs in population genetics is SmartPCA. SmartPCA was developed by Nick Patterson, Alkes L Price, Samuela Pollack, Kevin Galinsky, Chris Chang, Sasha Gusev, and David Reich. It is part of the package EIGENSOFT, which was technically developed only for Linux can be found here, but a Mac/Windows version, which can be found here, was also developed. SmartPCA will provide coordinates for samples depending on the principal component/eigenvector. An additional program in the EIGENSOFT package can be used to plot the data that is output by SmartPCA, though other programs can be used to plot the data as well.
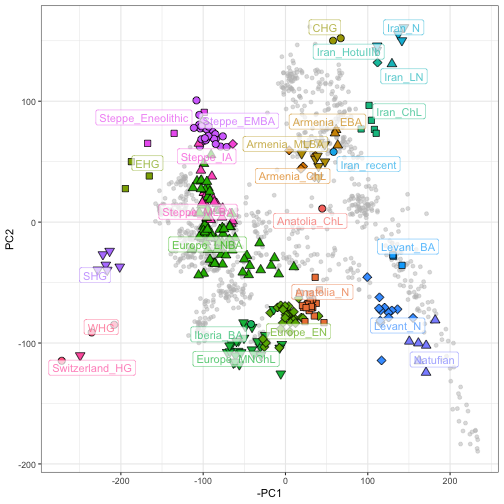
Image courtesy of Salvador Herrando-Perez and Christian Huber. This image displays what a SmartPCA graph can look like.
Another program used to generate PCAs (though it serves many other purposes as well) is PLINK. While PLINK is primarily known due its dataset manipulation and quality control faculties, PLINK also has a PCA function that, when used correctly, will yield results almost identical to SmartPCA. However, SmartPCA is more sensitive (meaning that coordinates will be more specific) and contains more features, such as automatic outlier removal. With adjustments to the output file (or adjustments to the commands used to graph results), PLINK PCA results can be graphed in the same manner as SmartPCA results. See more
Further Reading
Click here for more information about f-stastistics.
To learn more about any of the programs described, looking at the sources used in this article would be recommended.
Sources
GitHub - DReichLab/AdmixTools: Tools test whether admixture occurred and more
Programs Mentioned
EIGENSOFT (Original for Linux, contains SmartPCA)
EIGENSOFT (Experimental for Windows and Mac, contains SmartPCA) NOTE: In my personal experience, this version is far less powerful than the original Linux build and cannot handle larger (multiple GB) files. As a result, I reccommend that Windows and Mac users set up a Linux virtual machine if they want to utilize the full potential of EIGENSOFT.